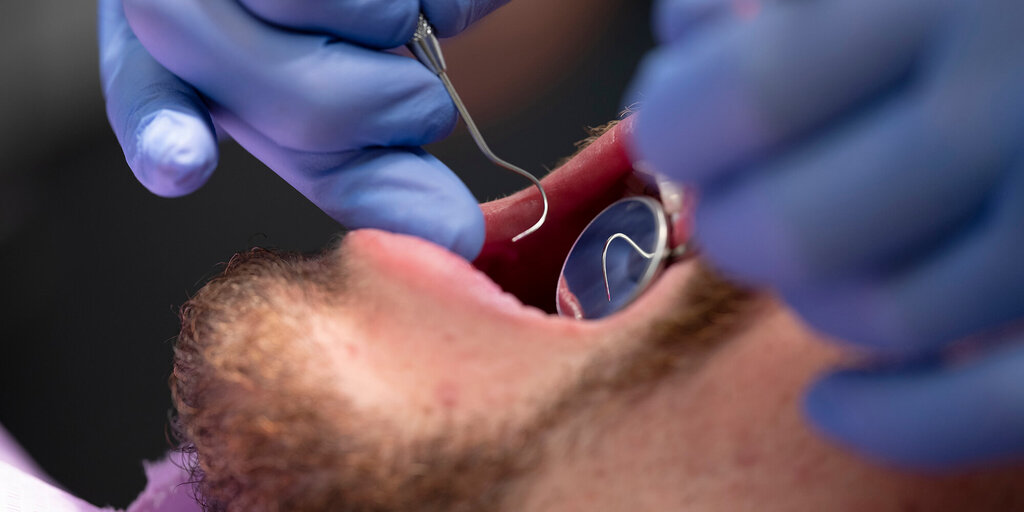
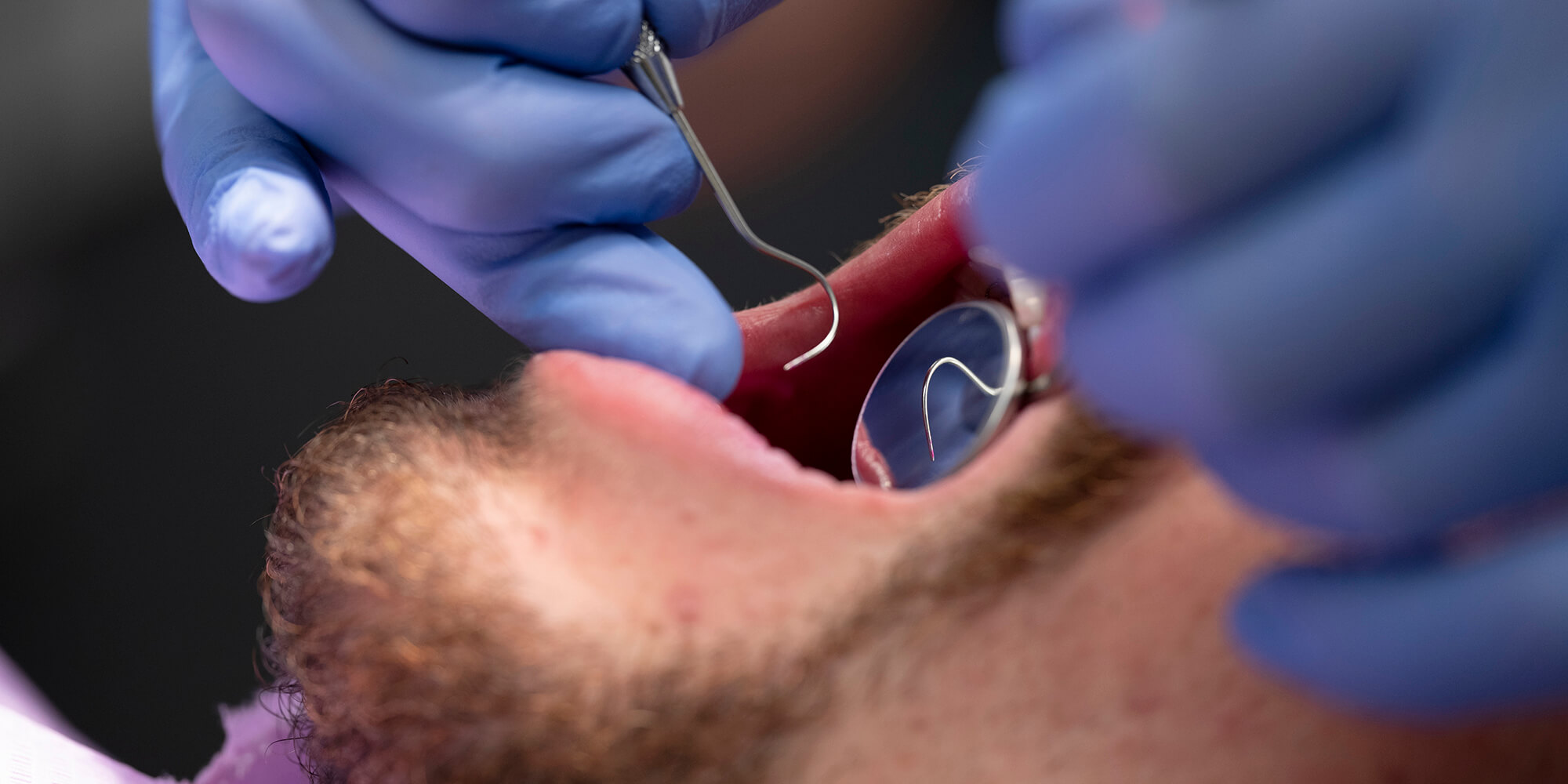
De ziekte van Von Recklinghausen of neurofibromatose type 1 (NF1) of maakt deel uit van een groep neurofibromatosen. Dit zijn neurocutane syndromen met huidafwijkingen en pathologie van het centrale zenuwstelsel. NF1 wordt onder andere gekenmerkt door neurofibromen, café-au-lait-vlekken, Lisch noduli en sproeten in de oksel en de lies. In het gelaat worden neurofibromen van de huid gezien. Intraoraal komen ook neurofibromen voor van het palatum, de tong en de gingiva, vaak leidend tot deformiteit van het gelaat en malocclusie. NF1 is een autosomaal dominante aandoening. Het gen voor NF1 is gelegen op chromosoom 17. Behandeling van de intraorale neurofibromen is moeilijk. Radicale verwijdering is niet goed mogelijk, contour corrigerende ingrepen leiden bij jonge individuen vaak tot snelle recidieven. Op volwassen leeftijd is recontouring, indien noodzakelijk en gewenst, mogelijk.
Von Recklingshausen´s disease (neurofibromatosis type 1 - NF1) is one of the neurofibromatoses and accounts for about 90% of all cases. Inheritance is autosomal dominant with about 30-50% of cases representing new mutations. Characteristic features for NF1 are six or more café-au-lait-spots, neurofibromas, Lisch nodules and axillary freckling. Oral manifestation consists of neurofibromas and intrabony lesions. Due to growth of the oral and facial neurofibromas maldevelopment of the facial skeleton and malocclusion are seen. Surgical correction in young individuals easily leads to recurrence. Contour corrected surgery in grown up individuals is possible.
| Auteur(s) |
J.A. Baart
J.M. van Hagen |
|---|---|
| Rubriek | Onderzoek en wetenschap |
| Publicatiedatum | 1 februari 2000 |
| Editie | Ned Tijdschr Tandheelkd - Jaargang 107 - editie 2 - februari 2000 ; 057-59 |
Er zitten geen programma's in het winkelmandje