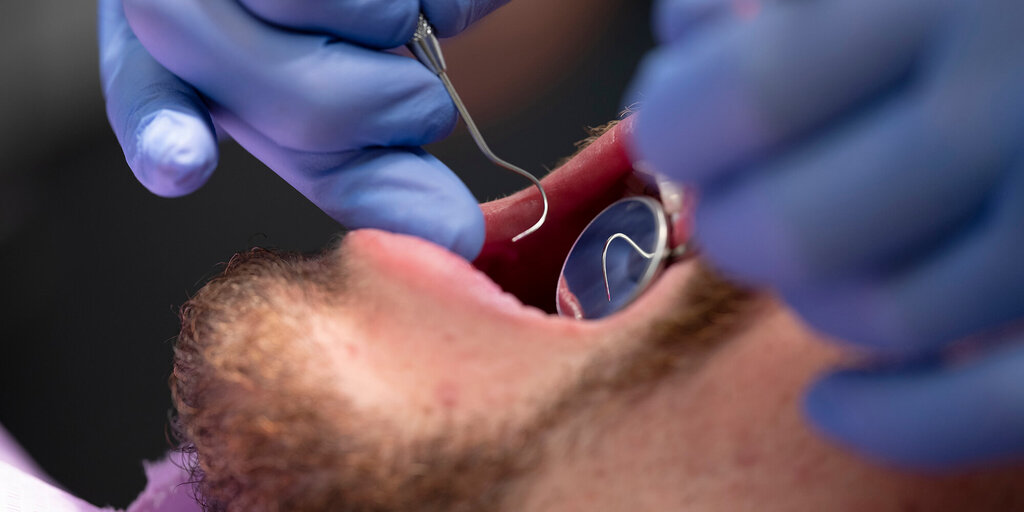
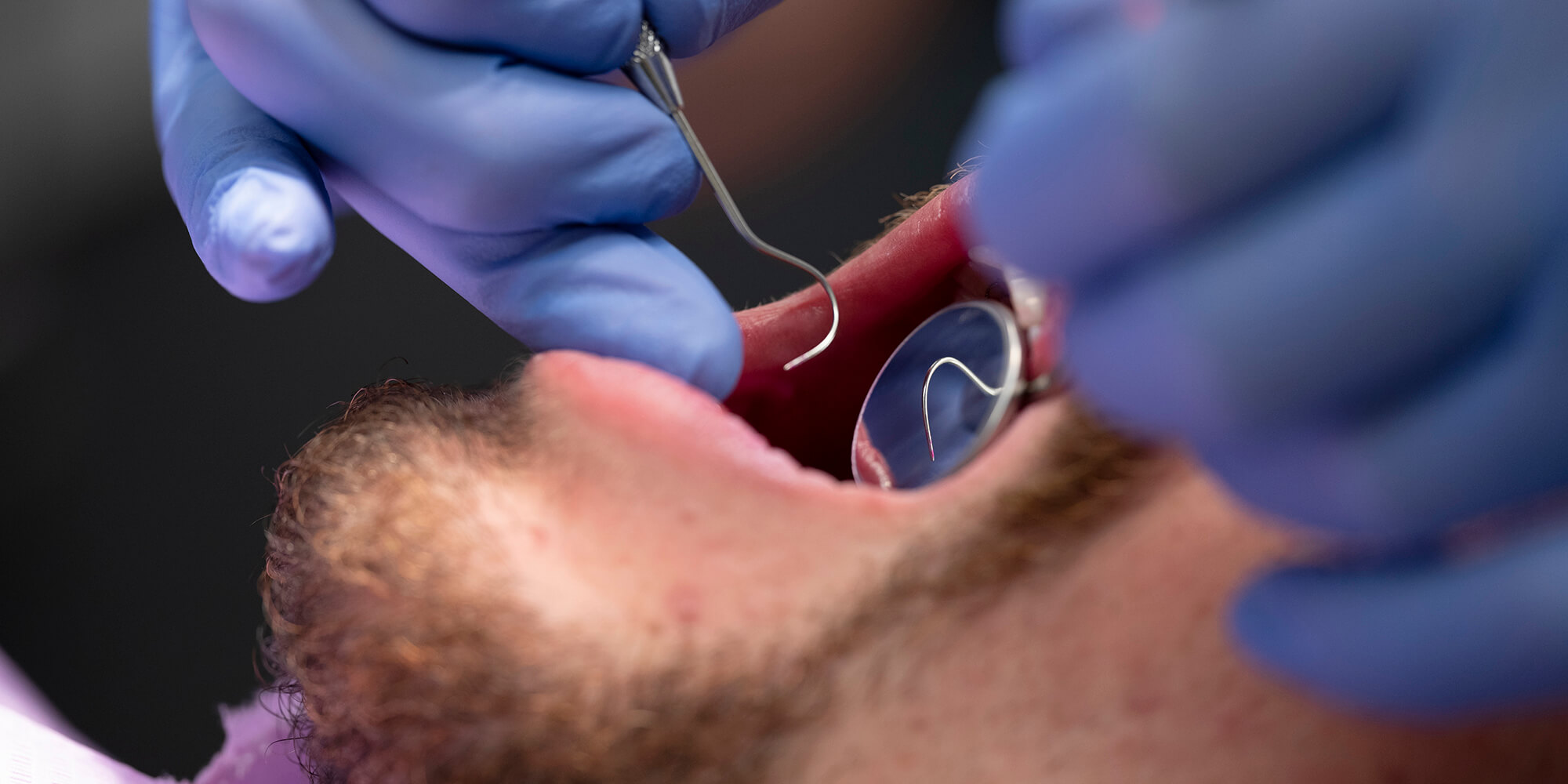
Fanconi-anemie is een recessieve erfelijke ziekte die wordt gekenmerkt door aplastische anemie en diverse andere klinische manifestaties, waaronder een predispositie voor acute myeloïde leukemie en plaveiselcelcarcinomen. De ziekte wordt veroorzaakt door mutaties in 1 van de 13 tot nu toe bekende genen die met dit ziektebeeld verbonden zijn. Deze genen coderen voor eiwitten die betrokken zijn bij reparatie van breuken in het DNA in de celkern. Plaveiselcelcarcinomen bij Fanconi-anemie ontstaan voornamelijk in de mond, maar ook in het anogenitale gebied, meestal op jonge leeftijd. Ze zijn moeilijk te behandelen omdat de patiënten extreem gevoelig zijn voor radiotherapie en chemoradiatie met cisplatine. Het vroegtijdig stellen van de diagnose en chirurgische interventie zijn daarom de beste remedie. Er zijn aanwijzingen dat het humane papillomavirus betrokken is bij het ontstaan van plaveiselcelcarcinomen bij patiënten met Fanconi-anemie, maar de onderzoeksresultaten zijn niet eenduidig. Vaccinatie tegen het humane papillomavirus en niet-invasieve screening naar premaligne slijmvliesafwijkingen moeten, in combinatie met frequente poliklinische controle en vroegtijdige chirurgische interventie, zorgen voor preventie en een betere prognose van deze tumoren.
Fanconi anemia is a recessively inherited disease, characterized by various anomalies, bone marrow failure, and a predisposition to acute myeloid leukaemia and squamous cell carcinoma. The disease is caused by mutations in one of the 13 currently known Fanconi anemia genes involved in the repair of DNA breaks. Squamous cell carcinomas arise chiefly in the oral cavity, but also in the anogenital region, mostly at relatively young age. Managing the carcinomas is difficult since the patients have are extremely sensitive to treatments such as radiation and chemoradiation using cisplatin. Therefore, early diagnosis and early surgical intervention are the best approaches. Human papillomavirus may play a role in the etiology of the oral squamous cell carcinomas, but current research results are inconsistent. Human papillomavirus vaccination and non-invasive screening for premalignant lesions, combined with frequent clinical examinations and early surgical treatment should improve the prevention and prognosis of these tumors in Fanconi anemia patients.
| Auteur(s) |
R.H. Brakenhoff
H. Joenje |
|---|---|
| Rubriek | Medisch |
| Publicatiedatum | 1 december 2007 |
| Editie | Ned Tijdschr Tandheelkd - Jaargang 114 - editie 12 - december 2007 ; 515-519 |
Er zitten geen programma's in het winkelmandje