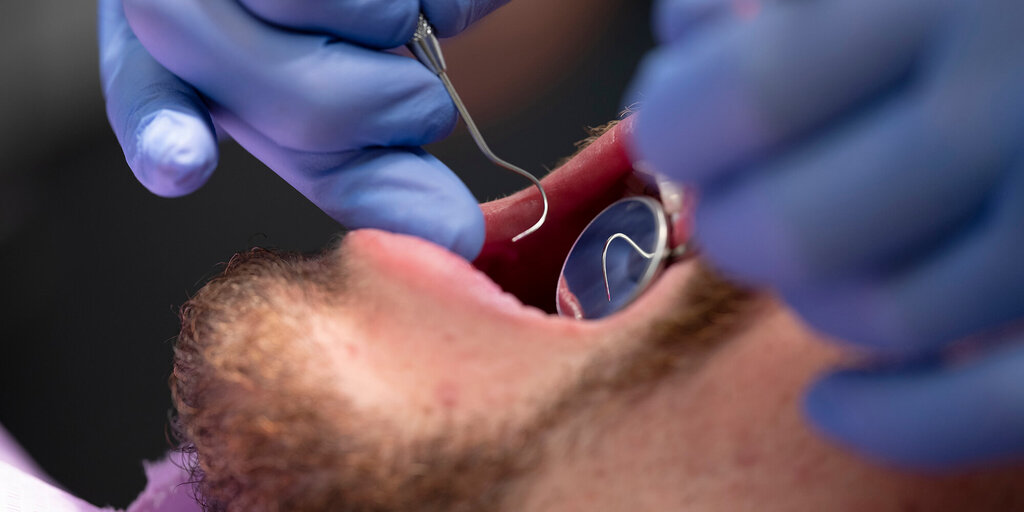
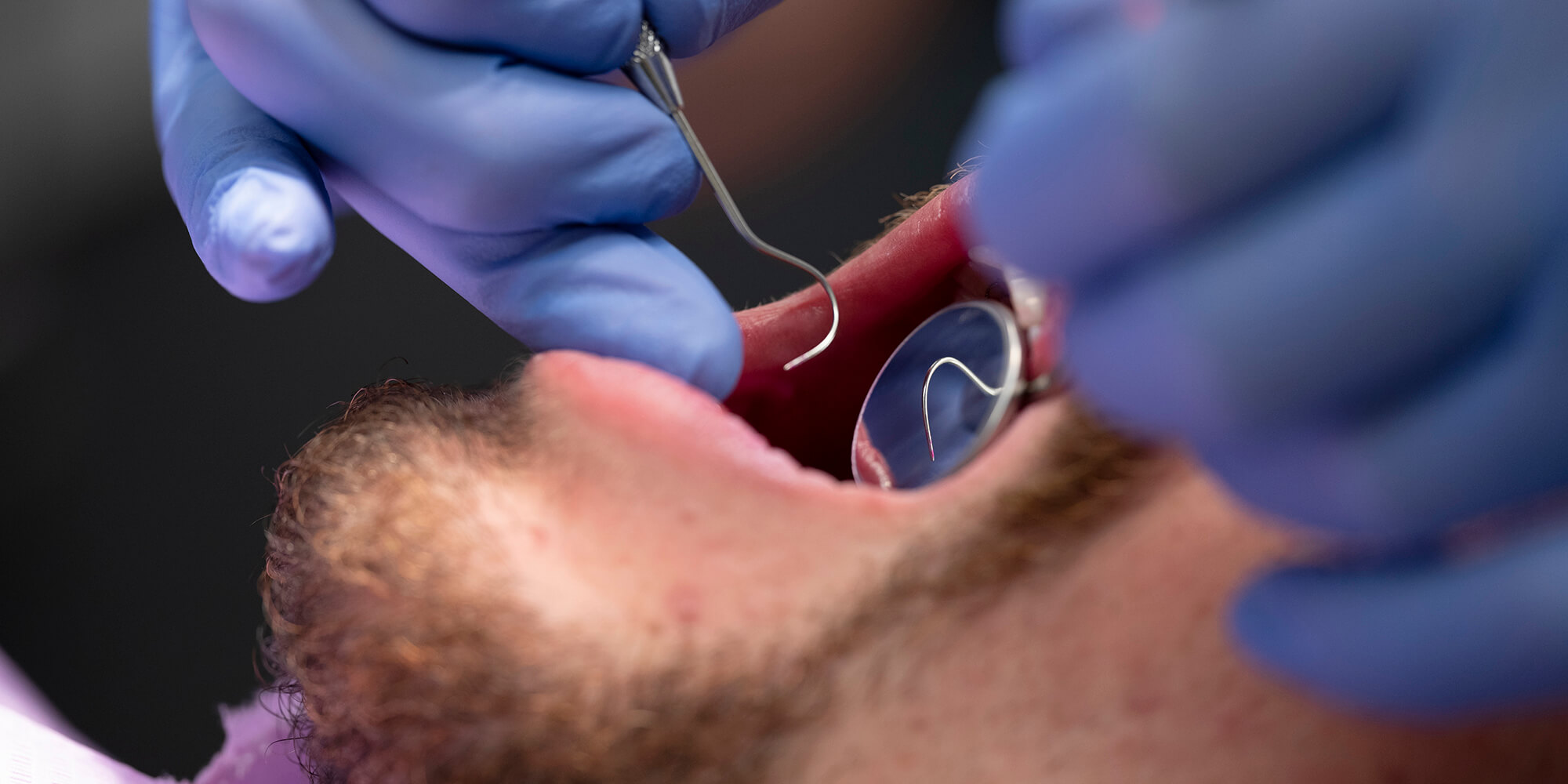
De ziekte van Rendu-Osler-Weber of heriditaire hemorragische teleangiectasien (HHT) is een multisystemische, autosomaal dominant erfelijke aandoening. Het ziektebeeld wordt gekenmerkt door multipele dysplasien van de bloedvaten op de huid en de slijmvliezen. Dit kan aanleiding geven tot recidiverende en fulminante bloedingen, waarvan een bloedneus de meest voorkomende is in deze patintenpopulatie. De arterioveneuze malformaties kunnen ook aanleiding zijn tot onder andere cardiale, pulmonale en cerebrale complicaties. Het ziektebeeld en een overzicht in de literatuur worden in dit artikel beschreven aan de hand van een casus.
Rendu-Osler-Weber disaese or hereditary hemorrhagic telangiectasia (HHT) is a multisystem autosomal dominant hereditary disorder. The disorder is manifested by multiple dysplasia of bloodvessels of the skin en mucous membranes. This results in recurrent and sometimes severe bleedings, of which epistaxis is the most common. Cardial, pulmonary and cerebral manifestations can be responsible for complications. A patient is presented with Rendu-Osler-Weber disease followed by a review of the literature.
| Auteur(s) |
L. Sys
F.J.A. van den Hoogen |
|---|---|
| Rubriek | Casuïstiek |
| Publicatiedatum | 1 september 2005 |
| Editie | Ned Tijdschr Tandheelkd - Jaargang 112 - editie 9 - september 2005 ; 336-339 |
Er zitten geen programma's in het winkelmandje